Major Research Interests
Our research focuses on the intersection of computational molecular biophysics and theoretical physical chemistry. We design and apply advanced computational approaches grounded in statistical mechanics and molecular thermodynamics to tackle challenging, interdisciplinary problems across chemistry, physics, and biology. The overarching aim is to gain a molecular-level understanding of how interactions and dynamics drive emergent functions and material properties. We employ molecular simulations, enhanced-sampling strategies, and machine-learning-based methods to investigate complex biomolecular and soft-matter systems — spanning small molecules, interfacial water, large proteins and enzymes, to self-assembled proteins. Our current research themes include (bio)molecular recognition and allostery, machine-learned discovery of collective variables for rare-event sampling, mechanistic studies of phase transitions, protein folding and aggregation, and AI-driven exploration of free-energy landscapes.
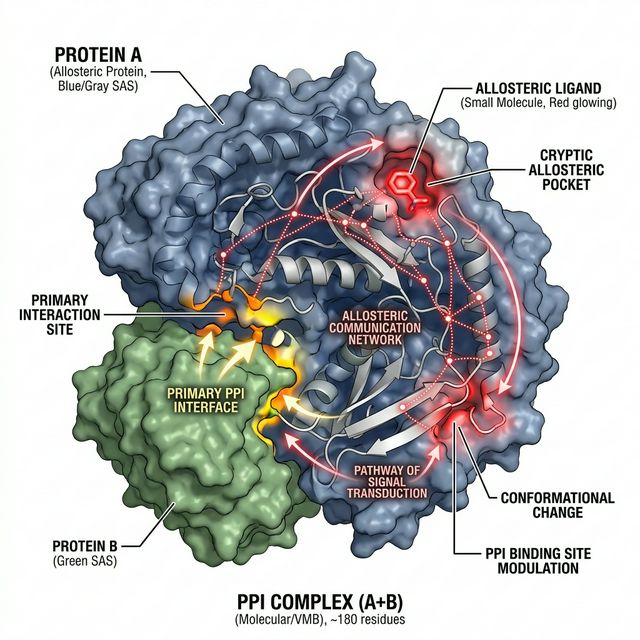
1. Protein Allostery & Interactions
Our lab investigates the intricate mechanisms of protein allostery and protein-protein interactions (PPIs). We employ advanced molecular dynamics simulations to understand how distant site perturbations, such as phosphorylation or ligand binding, dynamically regulate protein function. A key focus is uncovering druggable cryptic pockets and designing allosteric modulators for therapeutic intervention in complex systems like the PCSK9-LDLR complex and Rho GTPases.
Selected Publications
- Molecular mechanism of regulation of RhoA GTPase by phosphorylation of RhoGDI
- Phosphorylation induces altered protonation states and allosterically regulates Rac1–RhoGDI complex
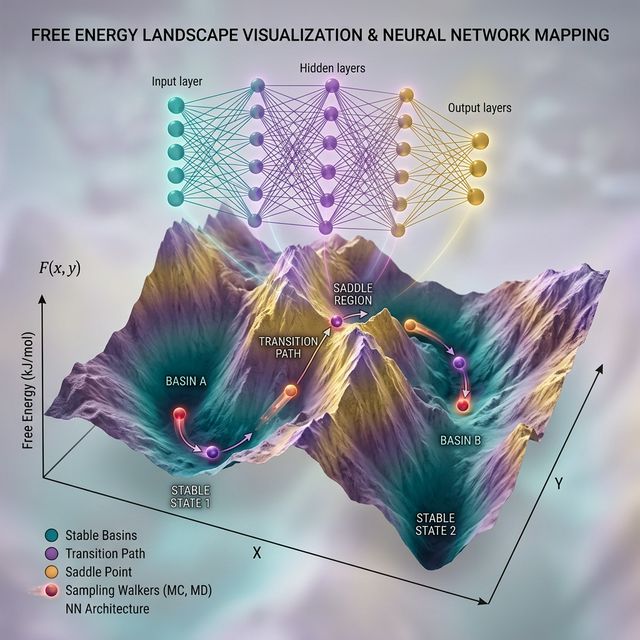
2. Machine Learning & Enhanced Sampling
We develop and apply cutting-edge machine learning and enhanced sampling techniques to overcome timescale limitations in classical molecular simulations. By integrating deep neural networks, variational autoencoders, and adaptive sampling strategies like PathGennie and WeTICA, we efficiently explore complex free energy landscapes. Our tools accurately predict rare event kinetics, ADMET properties, and phase transitions with remarkable computational efficiency.
Selected Publications
Show more!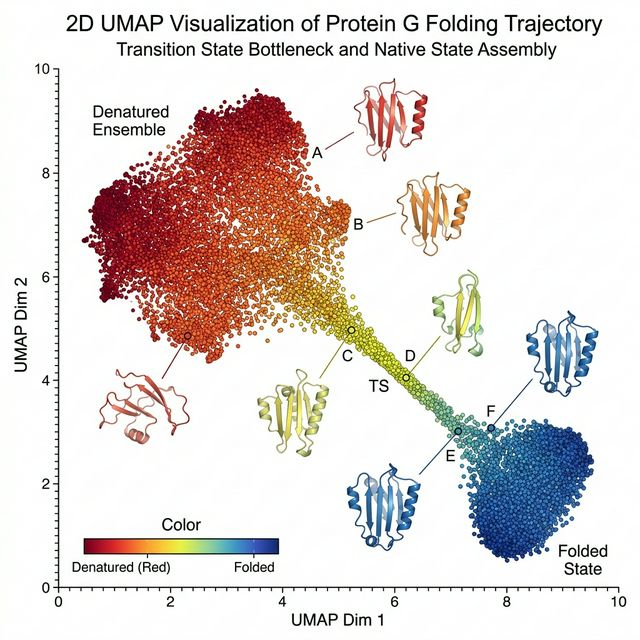
3. Biophysics & Conformational Dynamics
Understanding the fundamental biophysics of conformational dynamics is central to our research. We explore complex folding pathways, dimensionality reduction of conformational landscapes, and the mechanical stability of biomolecules. Through rigorous mixed-solvent simulations and structural analyses, we map interaction hotspots and elucidate how conformational diversity dictates molecular function and recognition.
Selected Publications
Show more!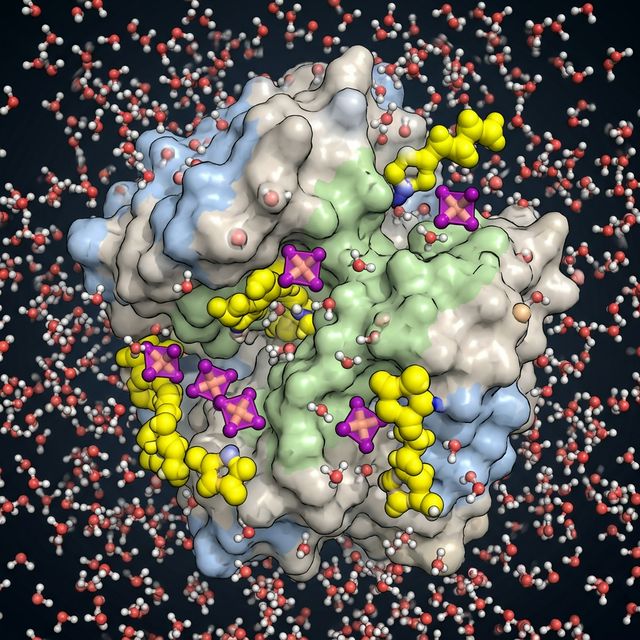
4. Solvation, Ionic Liquids & Phase Behavior
We investigate the microscopic origins of solvation thermodynamics and phase behaviors induced by complex solvent environments, including specialized ionic liquids and guanidinium salts. Our quantum chemical and molecular dynamics investigations reveal how specific ion-pairing propensities and surface-active molecules modulate protein (de)stabilization, hydrophobicity, and the delicate balance between protein-water and protein-protein interactions.