(Bio)molecular recognition and signaling are essential processes where proteins interact with specific molecules to regulate cellular functions. Molecular dynamics (MD) simulations provide an atomic-level, time-resolved view of these interactions, revealing how proteins recognize and bind to their targets through complementary shape, charge, and hydrophobicity.
In signaling, MD simulations track how binding events trigger conformational changes that propagate through the protein, leading to a biological response. These simulations are invaluable for understanding the dynamics of protein function, aiding in drug design by revealing key interaction mechanisms and potential targets for modulation.
Allosteric Modulation of PCSK9-LDLR Interaction
The protein PCSK9 negatively regulates LDL receptor (LDLR) recycling, leading to elevated plasma LDL levels. Targeting the PCSK9-LDLR interaction is promising for managing hypercholesterolemia; however, the large interaction surface poses challenges for small-molecule inhibitors.
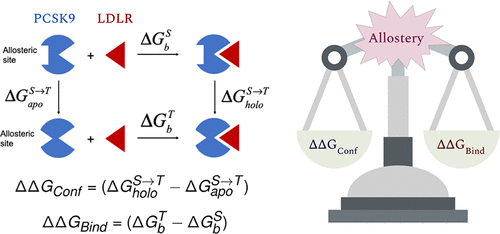
Using microseconds-long molecular dynamics (MD) simulations, we observed significant conformational changes in a distal loop (residues 211−222) of PCSK9 upon LDLR binding. These conformational shifts correlate with binding affinity, revealing potential allosteric hotspots for inhibitor design. Stabilizing apo-specific conformations of the loop may effectively reduce LDLR binding, providing a general strategy for identifying allosteric sites in proteins.
Phosphorylation Code in RhoGDI Regulation of Rho GTPases
Rho-specific guanine nucleotide dissociation inhibitors (RhoGDIs) regulate Rho GTPases by maintaining them in an inactive GDP-bound state. Phosphorylation of specific residues in RhoGDI selectively releases its GTPase partners, constituting a “phosphorylation code.”
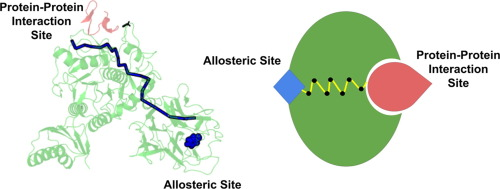
Using atomistic MD simulations of wild-type and phosphorylated RhoA-RhoGDI complexes, we demonstrate that phosphorylation induces structural changes in the positively charged polybasic region (PBR) of RhoA and the N-terminal region of RhoGDI, leading to reduced binding affinity. Hydrogen bond occupancy and energetic perturbation analyses reveal long-range allosteric signaling through rearrangements of salt bridges and hydrogen bonds. These results highlight the critical role of electrostatic interactions in mediating the phosphorylation code.
Allosteric Inhibition of Acetylcholinesterase (AChE)
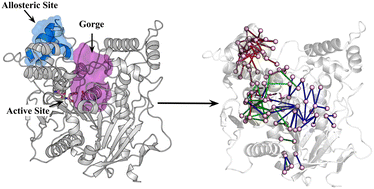
Acetylcholinesterase (AChE) is a key drug target for neurodegenerative disorders such as Alzheimer’s disease. Recent studies suggest certain antidiabetic drugs, like chlorpropamide (CPM), may act as noncompetitive inhibitors, though their binding sites and mechanisms were unknown.
Through MD simulations, we identified a previously unknown allosteric site in addition to the peripheral anionic site (PAS). Ligand binding induces population shifts in the catalytic triad, particularly HIS447, leading to enzyme deactivation. Inter-residue interaction network analysis elucidates the allosteric signaling pathways. These insights provide a foundation for rational design of novel noncompetitive AChE inhibitors.
References:
Pandey et al., Biochemistry 2023, 62, 5, 989–999
Das et al., Phys. Chem. Chem. Phys., 2024,26, 28894-28903 , 62, 5, 989–999
Sinha et al., J. Chem. Inf. Model. 2024, 64, 9, 3923–3932