Results in the paper¶
The Trails-MD paper demonstrates the framework on several systems beyond the
two runnable examples shipped in this repository (examples/AlaD/,
examples/AIB9/). This page summarizes those results; the underlying
configurations are not included in the repository, so these are
illustrative, not reproducible tutorials. For runnable, step-by-step
examples, see the Alanine dipeptide and
AIB9 tutorials.
Workflow overview¶
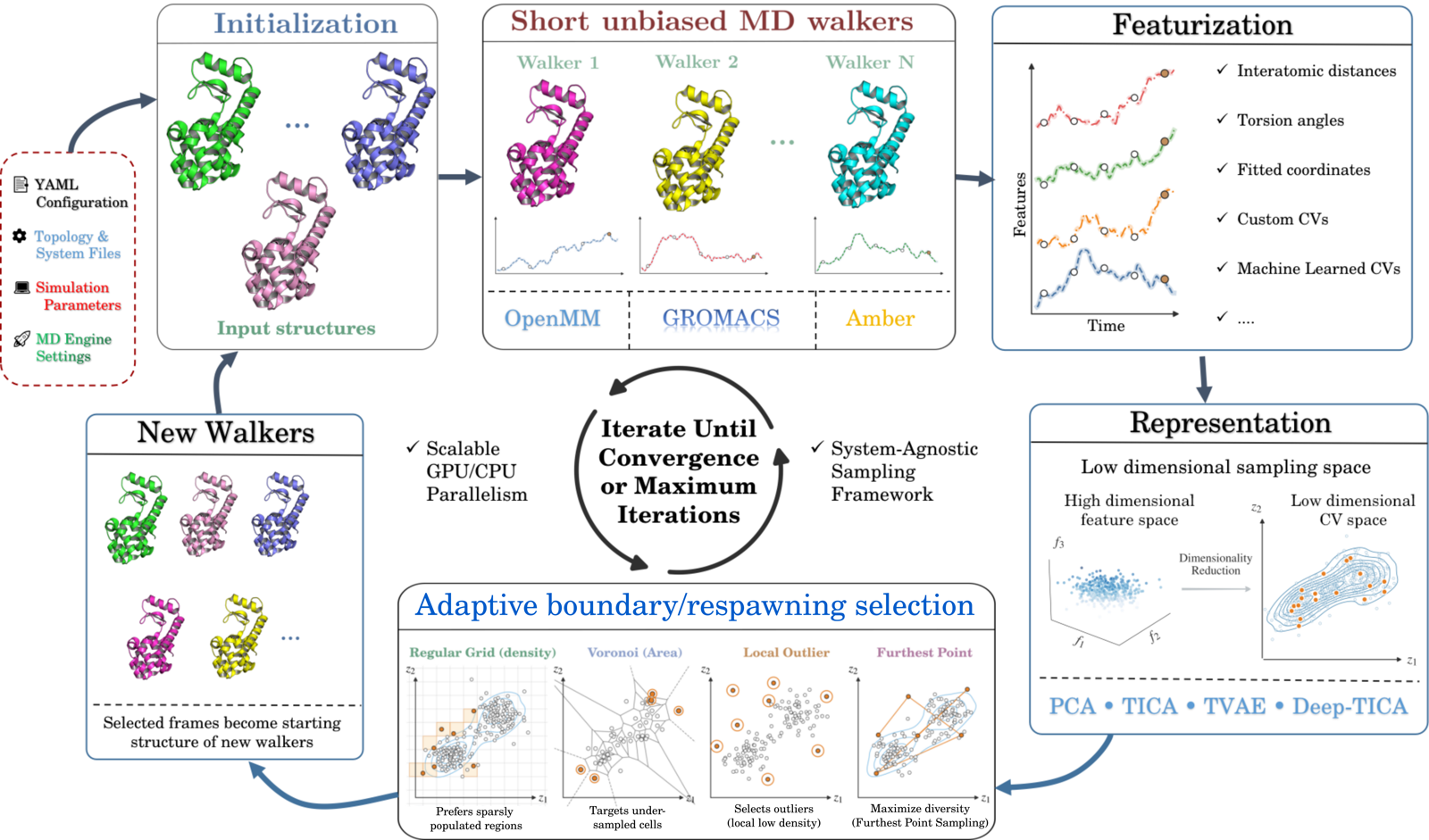
A YAML configuration defines the molecular system, MD engine, sampling space, spawning rule, and checkpointing options. Each iteration launches short unbiased walkers with OpenMM, GROMACS, or Amber, extracts features from the resulting frames, projects them into a fixed or learned CV space, and selects new starting frames with a density, Voronoi, local-outlier, farthest-point, MSM-guided, or weighted-ensemble spawner. Selected frames become the starting structures for the next batch of walkers, and the cycle repeats until convergence or a maximum iteration count.
CLN025 (chignolin) folding¶
Chignolin (CLN025) is a 10-residue peptide that folds into a stable β-hairpin, but — unlike alanine dipeptide — its folding is not naturally described by a single well-established low-dimensional coordinate. The paper compares three sampling spaces for CLN025: fixed physical CVs (Cα radius of gyration and Cα RMSD from the folded reference), and two learned 2D spaces built from Cα pairwise distances, TVAE and TICA.
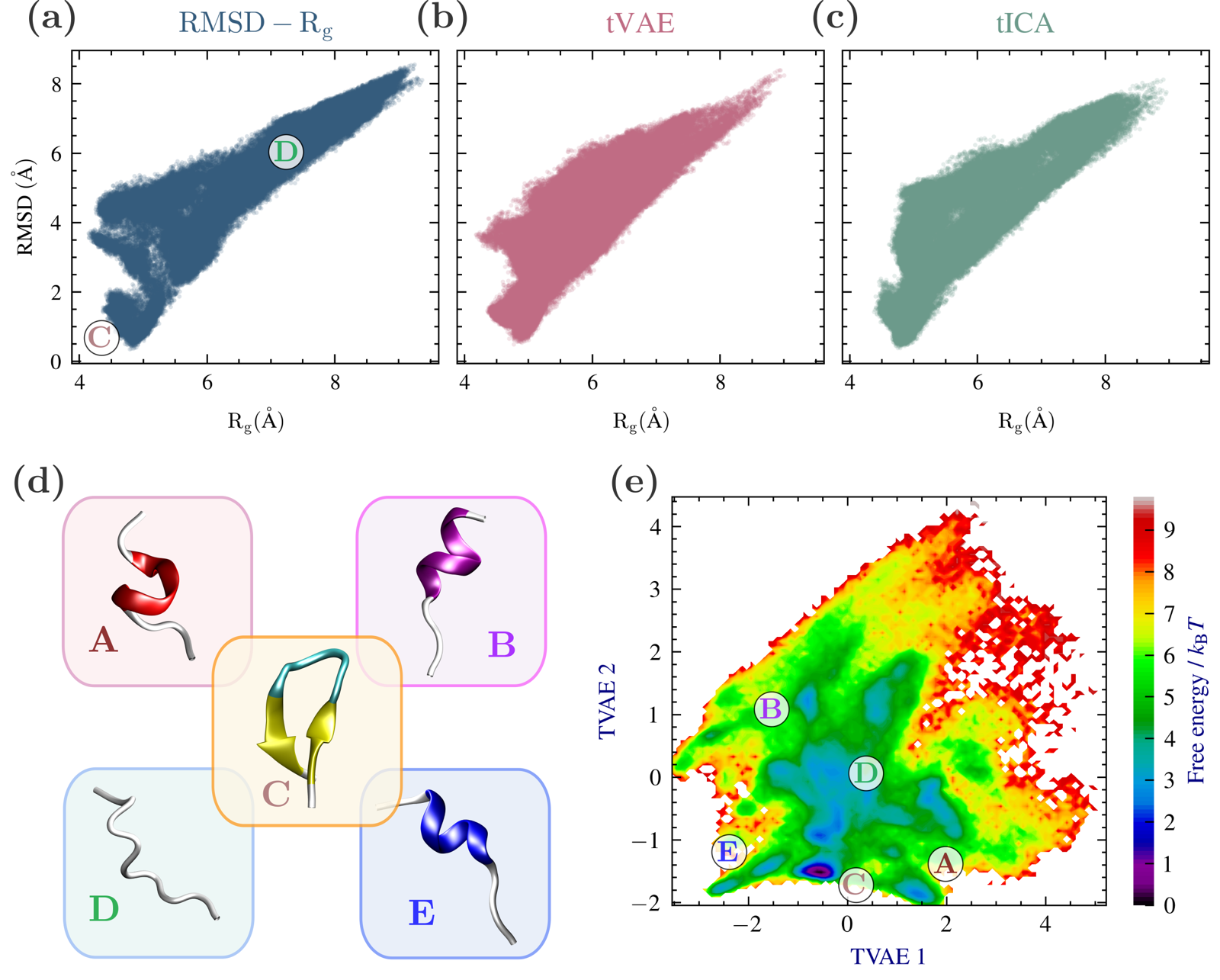
Conformations that overlap in the physical Rg–RMSD projection are more clearly separated in the learned TVAE space, illustrating the value of adaptive coordinate learning when an informative low-dimensional CV isn't known in advance.
AIB9: basin discovery is not a transition pathway¶
The AIB9 peptide makes a methodological point central to Trails-MD's design: the goal isn't only to visit the left- (L) and right-handed (R) helical basins, but to determine whether the sampled trajectories actually connect them.
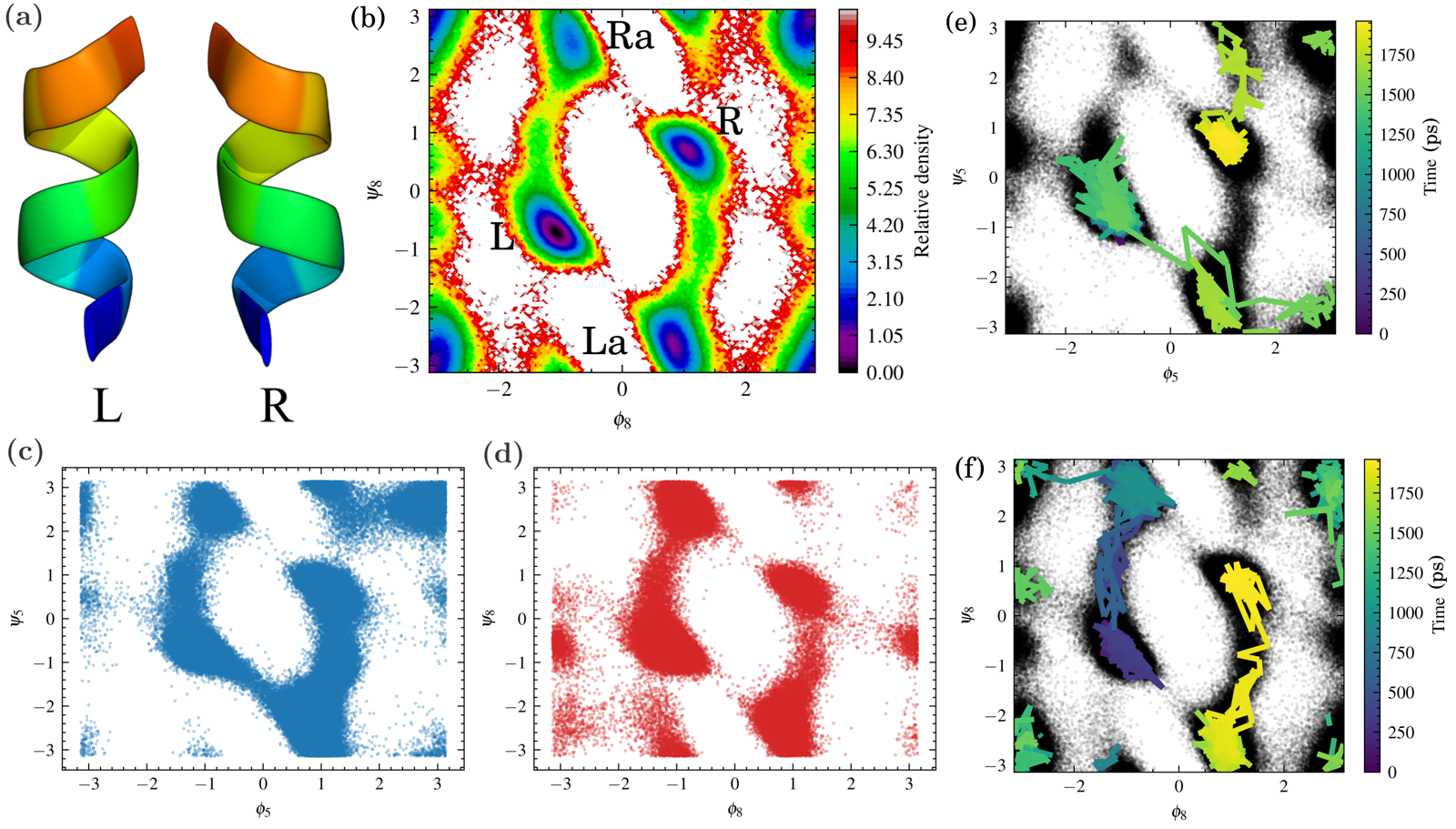
Several coordinate choices — fixed torsional angles, supervised LDA/TDA-based coordinates, and unsupervised PCA/TICA/TVAE spaces — all discovered both L and R basins, sometimes with high apparent coverage. But endpoint discovery didn't by itself establish a continuous dynamical route: lineage reconstruction showed that in several runs, the L- and R-like configurations belonged to different root trajectories.
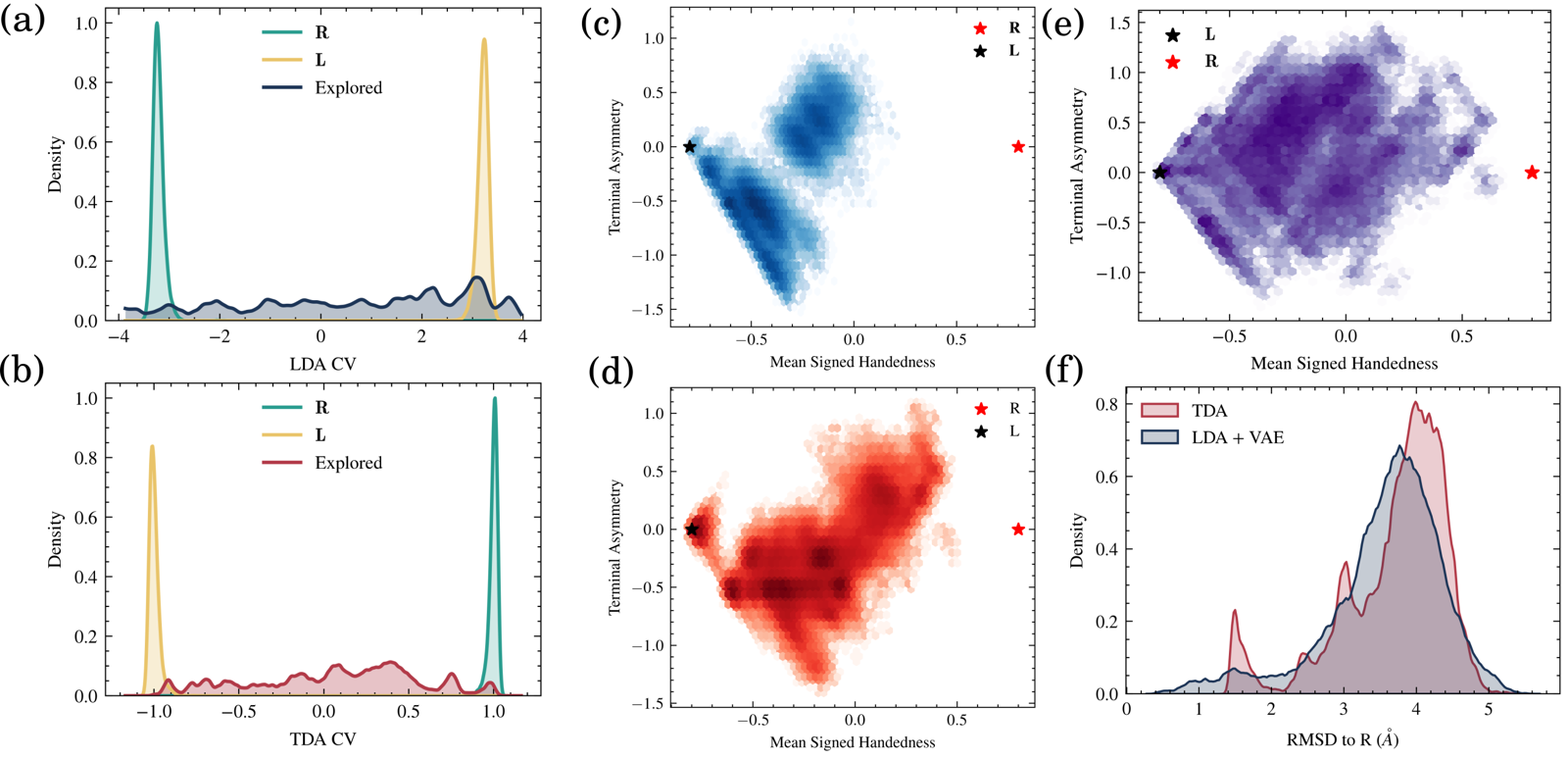
Combining an endpoint-discriminating coordinate with a structural latent
coordinate did recover a connected L-to-R pathway, traced from the ancestry
of the sampled frames. This is exactly what Trails-MD's parent-child lineage
tracking is for — see the runnable AIB9 tutorial for
the trails-md-path tool used to make this distinction.
Fast-folding proteins: Trp-cage and WW domain¶
Protein folding is a harder sampling problem than the low-dimensional benchmarks above, because common observables like RMSD or fraction of native contacts become increasingly degenerate away from the reference structure — many distinct unfolded or partially folded conformations can occupy similar regions of a low-dimensional projection.
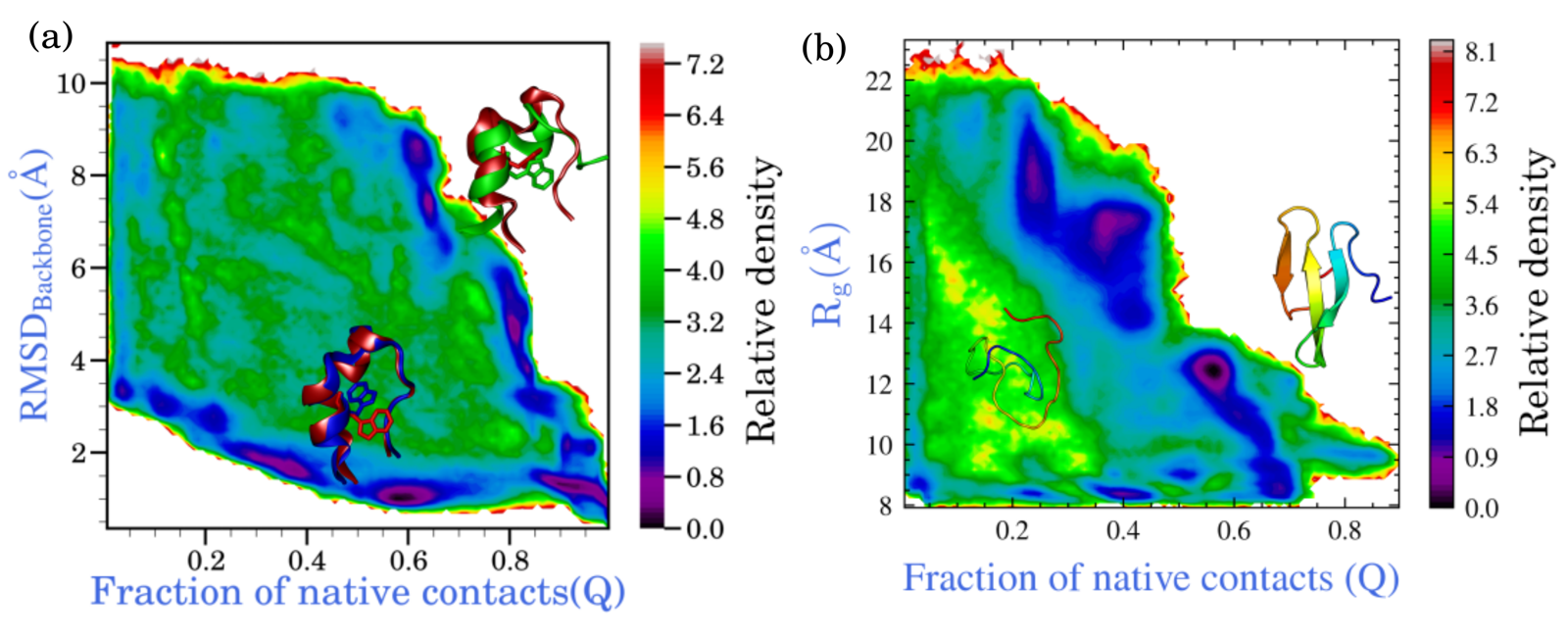
Trp-cage (2JOF, ~14 μs folding timescale) was sampled in a physically interpretable 2D space (RMSD from the folded structure vs. fraction of native contacts) using 25 walkers per iteration over 750 iterations, reaching the folded basin from an unfolded starting condition. The WW domain (~21 μs folding timescale) used a two-stage protocol: an unfolded ensemble was generated first (~50 iterations), a 3-component TICA space was trained on it, and a folding run in that learned space reached the folded basin within roughly 400 of 500 total iterations.
Ligand-unbinding tunnel mapping: OAMe-G2 and HSP90¶
Trails-MD can also map ligand-unbinding routes, not just conformational transitions. The paper tests two systems: the synthetic host–guest complex OAMe-G2, which has known "wet" and "dry" unbinding pathways, and inhibitor dissociation from the N-terminal domain of HSP90 (PDB 6EI5).
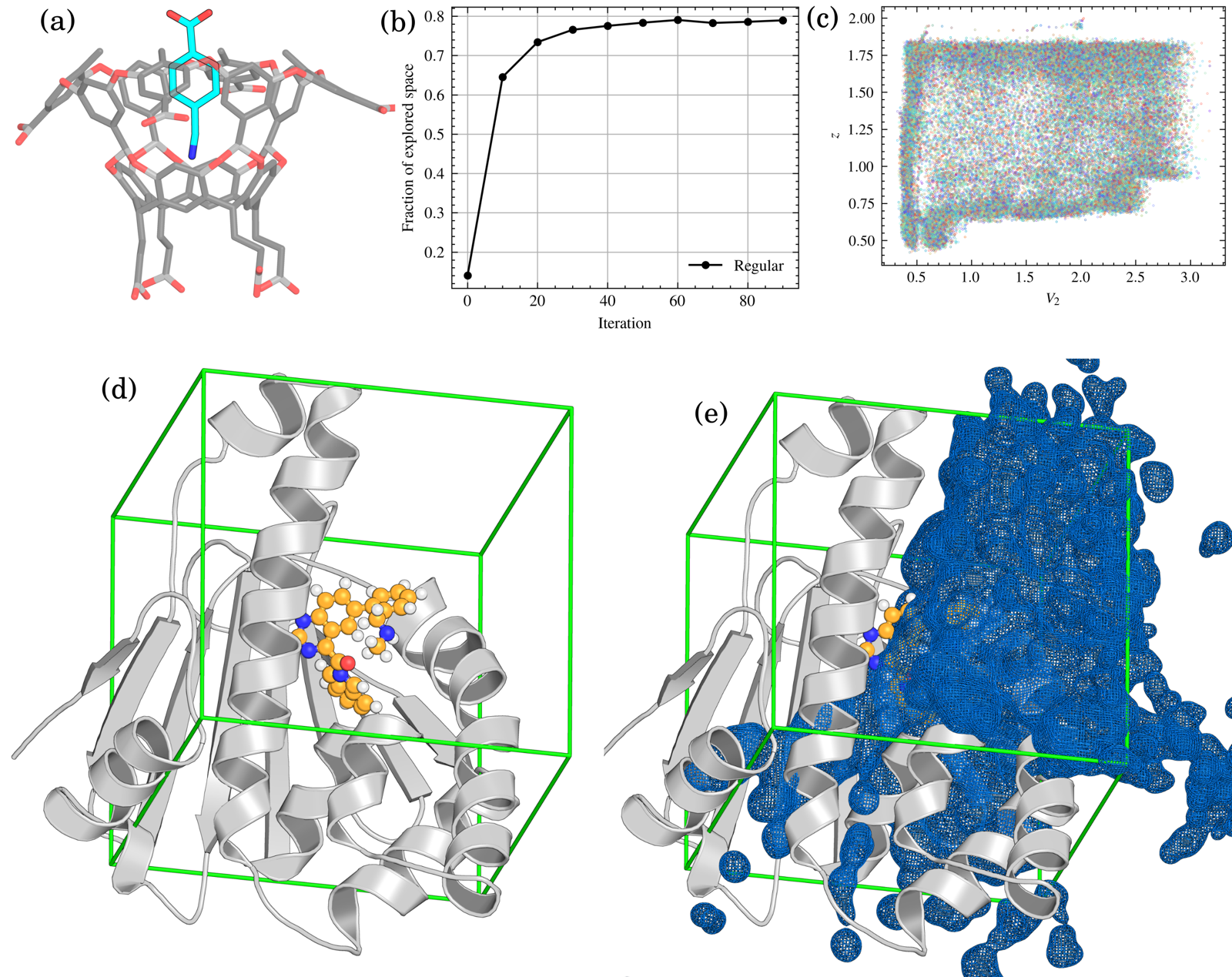
For OAMe-G2, Trails-MD progressively filled the projected unbinding space across multiple escape directions. For HSP90, sampled ligand positions were overlaid on the bound complex to map the accessible unbinding region and candidate exit tunnels — an exploratory tunnel-mapping stage that precedes more detailed kinetic or mechanistic analysis, rather than a substitute for it.
MSM construction from Trails-MD-seeded trajectories¶
Finally, the paper demonstrates the two-stage kinetic workflow described in MSM & kinetic seeding, tested on chignolin folding in explicit water (CHARMM27* force field, TIP3P water, 340 K).
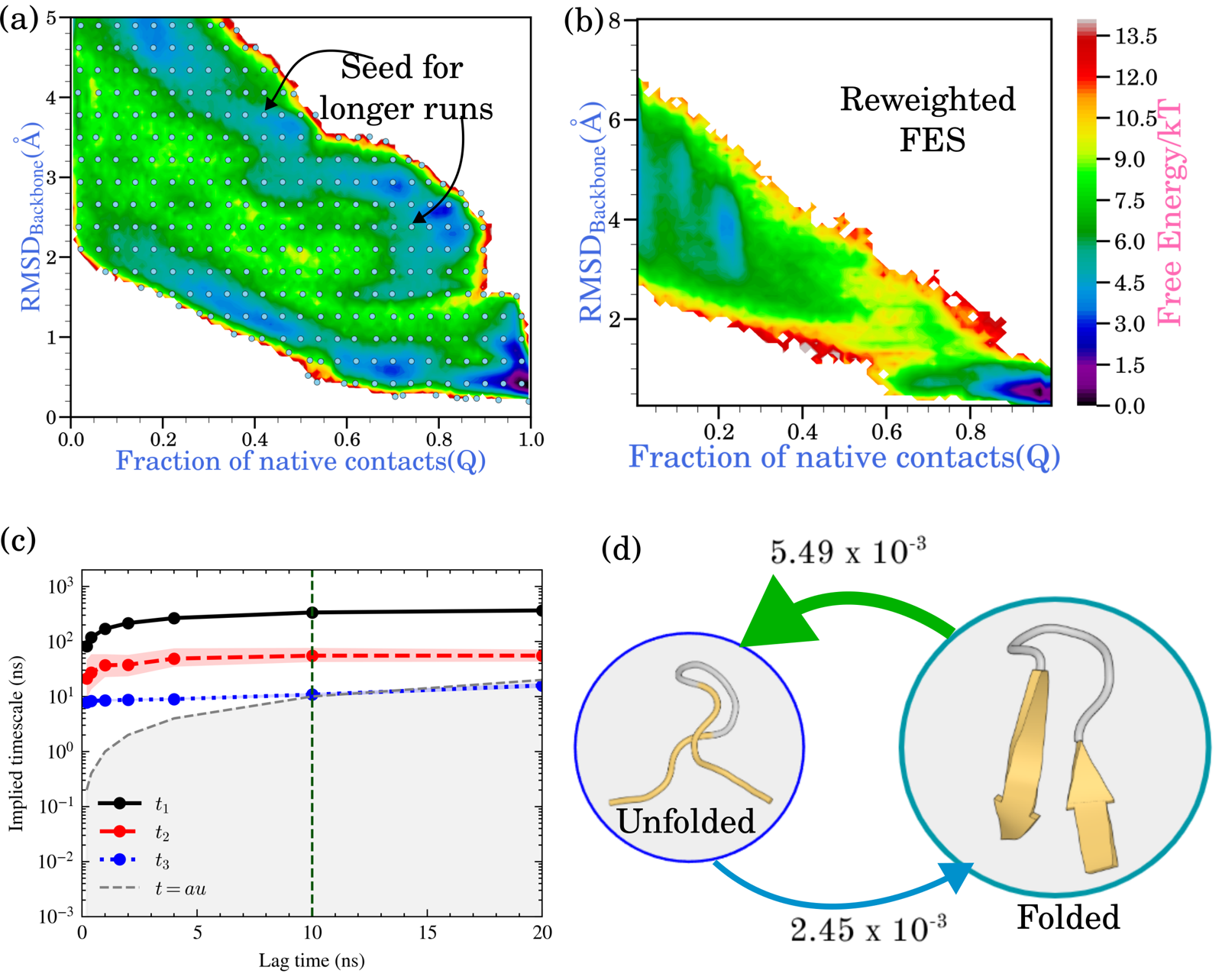
An adaptive campaign explored the RMSD–fraction-of-native-contacts space and, once coverage plateaued, 343 representative spawn points were selected from the discretized explored region. Each was used to seed a 50 ns production trajectory (≈17 μs aggregate), and an MSM was built from the resulting long trajectories with a 10 ns lag time chosen from implied-timescale convergence. A two-state coarse-grained model recovered folding and unfolding mean first-passage times of approximately 1.6 μs and 0.7 μs — consistent with chignolin's known microsecond-scale kinetics. This is a standard, external MSM-construction pipeline applied to Trails-MD-seeded data, not an in-loop MSM engine.